
Pobieranie materiałów kostnych do badań kopalnego DNA (źródło: 24K-PRODUCTION/SHUTTERSTOCK.COM)
Nasz indywidualny genom jest uważany za niezmienny, ale czasami pojawiają się mutacje. Ich identyfikacja może stanowić wyzwanie.
Janusz G. Zimowski
Bardzo często słyszymy, że otrzymany od naszych rodziców zestaw genów mamy dany raz na zawsze i że jest on niezmienny przez całe życie. W zasadzie to stwierdzenie jest prawdziwe, ale są od niego pewne odstępstwa. Pierwszym wyjątkiem – mimo działania systemów naprawczych, które mamy – jest wpływ szeroko rozumianego środowiska na nasz genom. Są to czynniki powodujące mutacje (światło UV, czynniki oksydacyjne, wiele związków chemicznych itp.), które mogą w poszczególnych komórkach naszego ciała prowadzić do zmian w sekwencji nukleotydowej DNA. Zmiany mogą być nieistotne i nigdy się o nich nie dowiemy, ale mogą też mieć poważne konsekwencje dla funkcjonowania komórek i prowadzić do procesów chorobotwórczych. W ten sposób tworzą się mutacje somatyczne, a więc takie, które powstają w komórkach budujących nasze ciało. Organizm stara się pozbyć uszkodzonych komórek, do czego wykorzystuje mechanizm programowanej śmierci komórki, czyli proces apoptozy. Niekiedy zmieniona genetycznie komórka wymyka się spod kontroli i staje się komórką nowotworową, zaczyna żyć życiem niezależnym od pozostałych komórek tego organizmu.
Mutacje
Innym wyjątkiem zmienności naszego genomu jest zbieranie informacji o tym, co spotkało organizm w czasie jego życia i „wpisywanie” tego w genom. Dzieje się tak przez chemiczną modyfikację DNA polegającą na metylowaniu niektórych odcinków bogatych w nukleotydy C i G (tzw. wyspy CpG). Proces ten jest najczęściej odwracalny i służy do regulacji działania wielu genów. On też może wymykać się spod kontroli organizmu i prowadzić do zmian patogennych. Powyższe modyfikacje, jeżeli nie dotkną komórek rozrodczych, nie są przekazywane potomstwu. Mogą jedynie wpływać na zdrowie osoby, w której genomie się pojawiają.
Inaczej przedstawia się sytuacja ze zmianami genetycznymi, które mogą pojawić się podczas powstawania komórek rozrodczych. Proces podziału komórek jest bardzo niedoskonały i może generować liczne błędy, tworząc poza prawidłowymi wadliwe komórki rozrodcze. Komórka zawierająca zmianę genetyczną, biorąc udział w zapłodnieniu, daje życie nowemu organizmowi i wprowadza do jego genomu ową zmianę. Część takich zmian wystąpi w sekwencjach niekodujących lub niemających znaczenia w procesach regulacji działania genomu i się nie ujawni (nie wpłynie na funkcjonowanie organizmu), część będzie miała znaczenie w połączeniu z odziedziczonymi po przodkach (wcześniej powstałymi) mutacjami i da efekt fenotypowy (zachorowanie), ale będą i takie, które dadzą efekt patogenny.
Oddzielną grupą zmian genetycznych jest wystąpienie mutacji w czasie embriogenezy, gdy dzielące się komórki różnicują się, tworząc klony komórek będących początkiem poszczególnych tkanek. Gdy zmiana sekwencji nukleotydowej DNA wystąpi w komórce, która jest jedną z pierwszych komórek klonu prowadzącego do powstania np. gonady, to zmiana ta będzie tylko wśród komórek powstającego organu. Pozostałe komórki tego organizmu będą – w znaczeniu genetycznym – prawidłowe i organizm rozwinie się bez widocznych zmian. Nosiciel/nosicielka takiej zmiany nie będzie mieć świadomości, że w jego/jej organizmie są zmutowane komórki. Ta zmiana genetyczna może ujawnić się dopiero w następnym pokoleniu i to pod warunkiem, że w zapłodnieniu weźmie udział zmutowana komórka rozrodcza.
Choroby genetyczne
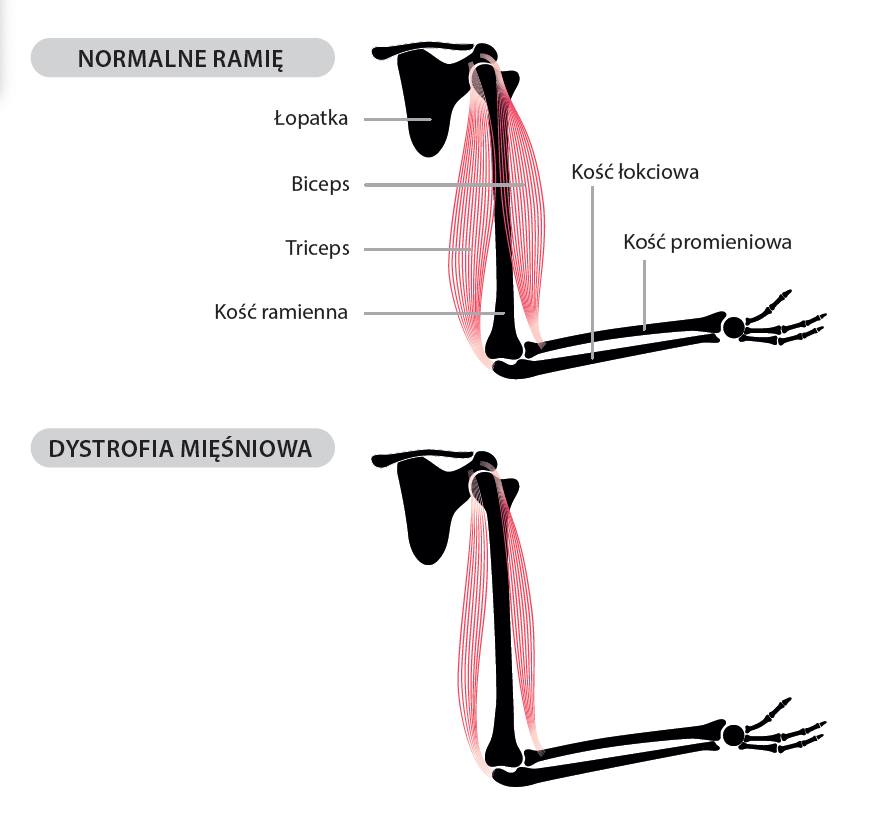
Wśród wielu poznanych chorób uwarunkowanych genetycznie znajduje się dystrofia mięśniowa Duchenne’a/ Beckera (DMD/BMD). Dziedziczy się ona w sposób recesywny sprzężony z płcią, co oznacza, że dotyczy chłopców, a kobiety pozostają zdrowe. Choroba w swojej ostrej postaci (DMD) ujawnia się – a właściwie jest rozpoznawana – przed końcem trzeciego roku życia, mimo że pewne dyskretne jej objawy mogą być zauważone wcześniej. Choroba powoduje postępujący i nieodwracalny zanik mięśni szkieletowych, prowadząc do unieruchomienia w 10. roku życia oraz do nieuchronnej śmierci w trzeciej dekadzie życia chorego. Podłożem tej choroby są zmiany w sekwencji nukleotydowej genu, położonego na chromosomie X odpowiedzialnego za kodowanie informacji o syntezie białka nazwanego dystrofiną.
Zmiany te powodują całkowity brak białka koniecznego do prawidłowego funkcjonowania komórek mięśniowych. Utrzymująca się stała częstość zachorowania (około 1 na 3500 żywo urodzonych chłopców) mimo bezpotomnej śmierci chorych mężczyzn świadczy o systematycznie nowo powstających tego typu mutacjach. Dodatkowo wyjątkowo ciężki przebieg choroby i brak terapii opartej na usunięciu przyczyny zachorowania powoduje wysokie zainteresowanie diagnostyką kobiet, które są nosicielkami, zwłaszcza w rodzinach dotkniętych DMD/BMD.
Diagnostyka molekularna nosicielstwa DMD/BMD polega na poszukiwaniu w genomie kobiety mutacji w genie dystrofiny. Oczywiście kobiety, mając dwa chromosomy X (mężczyźni mają jeden chromosom X i jeden chromosom Y), posiadają dwie kopie tego genu, a nosicielki DMD/BMD jedną uszkodzoną, a drugą prawidłową. Produkowane białko z nieuszkodzonej kopii genu zapewnia kobietom nosicielkom prawidłowe funkcjonowanie komórek mięśniowych. Niestety, kobieta nosicielka DMD/BMD ma wysokie, bo 50-proc. ryzyko urodzenia syna cierpiącego na tę chorobę.
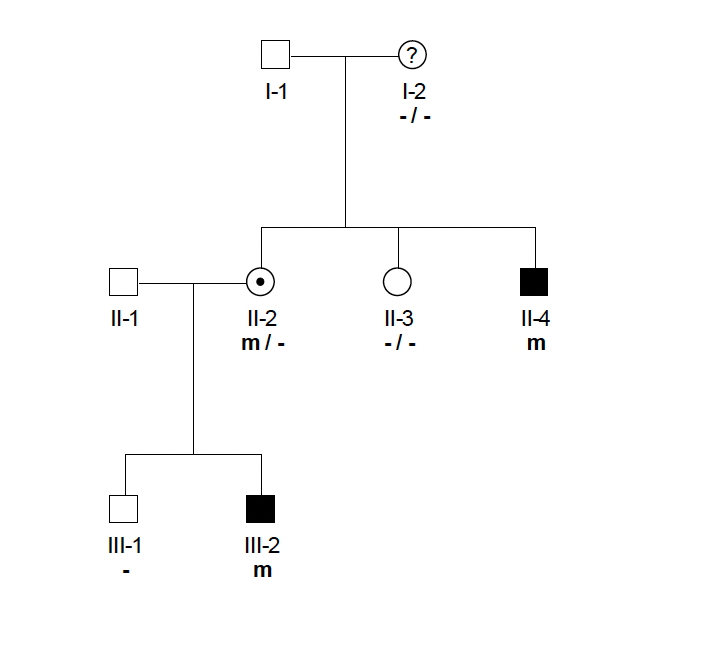
Kobieta w rodzinie dotkniętej dystrofią mięśniową Duchenne’a/Beckera, w której np. chory jest brat i syn, jest na pewno nosicielką choroby, a mutację odziedziczyła po swojej matce. Pojawienie się pierwszego zachorowania w rodzinie (brak informacji o innych chorych krewnych w rodzinie matki) stawia pytanie, czy matka chorego syna ma w swoim genomie ukrytą mutację i jest zagrożona ryzykiem urodzenia kolejnego dziecka, które będzie mieć to schorzenie, czy też jest to przypadek spowodowany nowo powstałą mutacją i ryzyko wystąpienia kolejnego zachorowania jest bardzo niskie, równe populacyjnemu. Uwzględniając stałą częstość wystąpienia DMD/BMD i bezpotomną śmierć chorych osób, teoretyczne prawdopodobieństwo nosicielstwa przez matkę (u syna dotkniętego chorobą) wynosi 66 proc. Oznacza to, że 33 proc. matek chorych chłopców nie jest obarczona nosicielstwem choroby. Rozstrzygającą odpowiedź na to pytanie może dać badanie molekularne.
Diagnostyka
Analizę rozpoczyna się od zidentyfikowania mutacji u chorego chłopca, by potwierdzić rozpoznanie kliniczne postawione przez lekarza. W dalszej kolejności prowadzi się badania nosicielstwa u kobiet z nim spokrewnionych. Badanie nosicielstwa tak jak i badanie molekularne chorego wykonuje się z użyciem DNA wyizolowanego z komórek krwi. Przyjmuje się, że są one reprezentatywne dla wszystkich komórek badanego organizmu. Znalezienie w tak skonstruowanym badaniu mutacji u chorego i jego matki daje jednoznaczną informację – pewny status nosicielstwa. Inaczej jest w przypadku nieznalezienia mutacji zidentyfikowanej u chorego syna.
Zwykle jest to interpretowane jako wykluczenie nosicielstwa, jednak taki wniosek może być obarczony błędem wynikającym z użycia do badania DNA materiału genetycznego niepochodzącego z komórek rozrodczych badanej osoby. Kobiety, u których w badaniu molekularnym nie wykryto mutacji genu dystrofiny, a które rodzą kolejne dziecko z mutacją powodującą DMD/BMD, mają tzw. mozaikowość germinalną, czyli klony komórek zmutowanych wśród komórek prawidłowych. Możemy taką sytuację określić mianem ukrytego nosicielstwa. Szacuje się, że częstość mozaikowości germinalnej wśród kobiet, u których wykluczono nosicielstwo dystrofii mięśniowej Duchenne’a/Beckera, wynosi powyżej 10 proc.
Przedstawione zjawisko mozaikowości germinalnej stanowi istotny problem w poradnictwie genetycznym, uniemożliwiając pewne wykluczenie statusu nosicielstwa. Z tego powodu kobiety, które urodziły syna dotkniętego dystrofią mięśniową Duchenne’a/Beckera i u których w badaniu molekularnym ich genomu nie wykryto mutacji powodującej chorobę, należy zawsze poinformować, że mają podwyższone ryzyko urodzenia syna dotkniętego DMD/BMD lub córki nosicielki tej choroby. ■
Wersja drukowana tego artykułu ukazała się w numerze 2/2023 (74) popularnonaukowego kwartalnika „Academia: Magazyn Polskiej Akademii Nauk” [ściągnij pdf].
